1995, Biochemistry, 60, 8, 923-928.
GeneBee-NET: Internet-Based Server for Analyzing Biopolymers Structure
L. I. Brodsky,1,2 V. V. Ivanov,1 Ya. L. Kalaydzidis,3 A. M. Leontovich,3
V. K. Nikolaev,1 S. I. Feranchuk,1 and V. A. Drachev3
'
Small Scientific Manufacturing Enterprise Gendalph, Moscow, 119899Russia; E-mail: libro@cdrm.genebee.msu.su
2
To whom correspondence should be addressed3
Belozersky Institute of Physico-Chemical Biology, Lomonosov MoscowState University, Moscow, 119899 Russia; fax: (7-095)939-39-89; E-mail: libro@genebee.msu.su
Submitted December 21, 1994; revision submitted April 5, 1995.
This work describes a network server for searching databanks of biopolymer structures and performing other biocomputing procedures; it is available via direct Internet connection. Basic server procedures are dedicated to homology (similarity) search of sequence and 3D structure of proteins. The homologies found could be used to build multiple alignments, predict protein and RNA secondary structure, and construct phylogenetic trees. In addition to traditional methods of sequence similarity search, the authors propose "non-matrix" (correlational) search. An analogous approach is used to identify regions of similar tertiary structure of proteins. Algorithm concepts and usage examples are presented for new methods. Service logic is based upon interaction of a client program and server procedures. The client program (GeneBee for IBM PC) allows the compilation of queries and the processing of results of an analysis. Without the client program it is possible to send queries via E-mail and examine results in text form. The server is available via E-mail as SERVE@INDY.GENEBEE.MSU.SU and also as WWW-server WWW.GENEBEE.MSU.SU.
KEY WORDS: mathematical and statistical biology, biological sequences databanks, macromolecular algorithms, sequence homology, sequence alignment, RNA secondary structure, protein secondary and tertiary structure, protein structure alignment.
In the 70s and 80s the size of the biopolymer sequence and structure databanks was small enough to keep them on a personal computer. Now this is impossible due to the enormous size of these databanks and because it is necessary to update them daily. Compact disk (CD-ROM) releases of databanks by NCBI and EMBL do not solve the problem because they appear with substantial delays [1]. The size of modern databanks often requires for their processing computing resources which are not available at the individual working desk of a researcher.
In recent years many computer centers were established in the West offering biocomputing services via direct Internet access [2-4]. Free services provided by these centers are mainly keyword and homology searching of
sequence databanks. It is assumed that for more detailed analysis the researcher can use one of the commercially available packages (e.g., GCG [5] which can be found in most European and American universities). Frequently, these packages are also accessible on the network [6], but with access for outside users restricted by the institute administration.
Scientists from Russia and other CIS states are in a position of disadvantage. The relatively high cost of telecommunications in Europe and America limits their access to international biocomputing centers even if the centers provide their services free of charge. Therefore, Belozersky Institute of Physico-Chemical Biology, together with SSME Gendalph and with financial assistance from the Russian Foundation for Fundamental Research, created a national computer center for support of bio-molecular research programs. Connected to Internet, this center enables network access to its resources, processes form-based queries, and performs specific biocomputing tasks for individual researchers.
Programs from the package GeneBee for IBM PC with transputing complex [7, 8] were utilized for some of the servers computational procedures and new programs were written in part for SGI graphics workstation and for the transputing complex. This paper describes the design of the algorithms on which these programs are based.
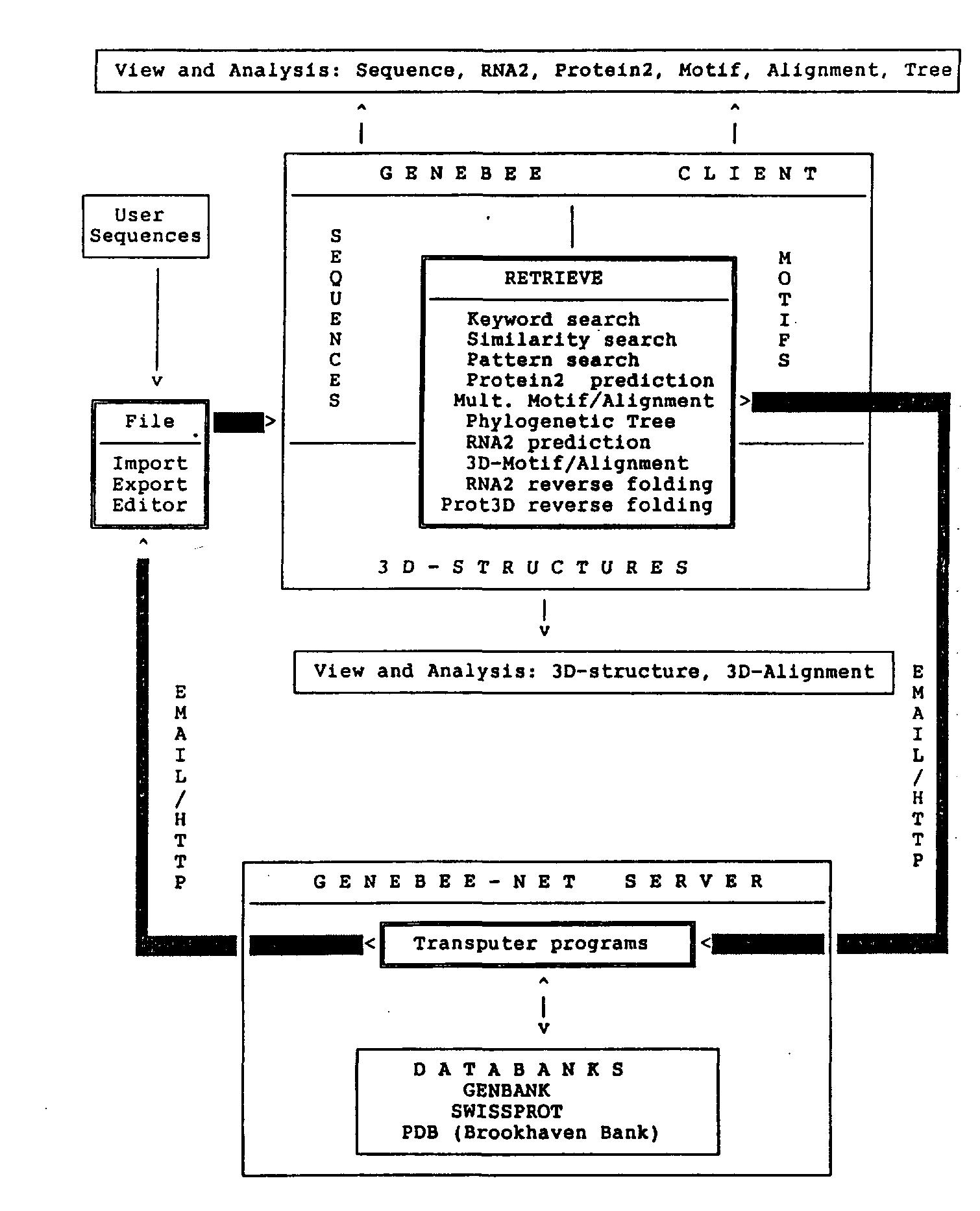
Fig. 1. Schematic representation of GeneBee-NET server and GeneBee client interaction. Either E-mail or HTTP protocol (if TCP/IP connection can be established between server and client) can be used for interfacing with the client program. Installation of the GeneBee client program provides the user with the interface and programs for query reply data processing and visualization (View and Analysis blocks). RETRIEVE block lists all kinds of server queries. While working with WWW-server, the user compiles a query interactively with the help of the server menu which is displayed on the user's computer by the WWW browser (Mosaic, Netscape, or Lynx).
SERVER ORGANIZATION
The GeneBee-NET server stores in special format and regularly updates the following databanks: nucleotide sequences (GenBank); protein sequences (SwissProt); 3D-structure of biopolymers (PDB).
The nucleotide sequences databank is daily automatically updated every 12 h. Data are downloaded via FTP from the National Center for Biotechnology Information of the National Institute of Health (NCBI-NIH) and archived to single database on the central computer of the complex—Indy Silicon Graphics workstation. Each new full release of GenBank is also automatically downloaded via FTP and archived, thus synchronizing accumulated updates. The two other databanks (SwissProt and PDB) are updated automatically as soon as new releases are made available on the FTP sites.
The interaction of end users with GeneBee-NET is arranged as a client-server system. The users computer (IBM PC compatible) can be equipped with the client program, allowing to compile server queries in graphics and pseudographics modes, make connection with the server via electronic mail or by HTTP (HyperText Transfer Protocol), and collect and interpret results.
Queries can be created using the client program or a sample form file. Figure 1 shows a schematic representation of the GeneBee-NET server and the GeneBee client interaction.
PROCEDURE ALGORITHMS
Some of the algorithms for server procedures have been published: sequence or alignment homology database search [7], multiple alignment of sequences [8], construction of phylogenetic trees [7, 9, 10]. We will review the unpublished procedures.
1. Pattern (Consensus) Search
The format of the search pattern coincides with the Prosite format (database of functionally meaningful protein patterns [11]). First, all fragments are found which fit the pattern at least in some conditions and then a fixed number of the best hits is selected. To do this, function F is calculated as follows. All pairs of positions (i, j) of the analyzed sequence in which one of the letters permitted for the pattern is located are marked. If for any of these pairs the distance between two positions meets the restrictions imposed by the pattern, then the value of function F in the corresponding position is incremented by one. After all pairs of sequence positions are processed we should get all values of F(i) function (i, position number). Local maxima of this function will show putative searched pattern sites (pattern could have "errors"). Next, the Dothelix [12, 13] algorithm is applied to separate sequence regions conforming to the pattern most strictly.
2. Correlationally Linked Fragments: Databank Search and Alignment
Databank homology search usually selects statistically significant MOTIFS (local alignments of sequences without insertions/deletions). Motif significance is estimated by its POWER [12, 13], specified by the equation:
Summing in the divisor is carried out by motif position number i; W is letter (residue) substitution weight matrix; L is length of the motif (number of positions); M and s are mean and root mean square deviation for W matrix elements, taking into account frequency of letters in the first and second sequences; ai and bi are letters in the i-th position of the motif.
Searching for statistically significant motifs with the help of the residue substitution weight matrices imposes a problem of selecting a "correct" matrix (or matrices) [14-17]. In practice, several essentially different types of matrices are used (identity matrix, Dayhoff matrices, matrices of residue physicochemical properties: hydropho-bicity, polarity, secondary structure propensity, etc.). It should be noted that, in fact, every substitution weight matrix determines the type of statistical dependence of letters in a motif, selected by the Eq. (1) criterion [17]. Hence, the idea arises to look not for motifs associated with a certain matrix or set of matrices, but for all motifs which demonstrate any kind of statistical dependencebetween their fragments. This is achieved by introducing a specific estimate—"correlational power"—which shows the statistical conformity of transition inside the motif from the letters of one sequence to letters of another one:
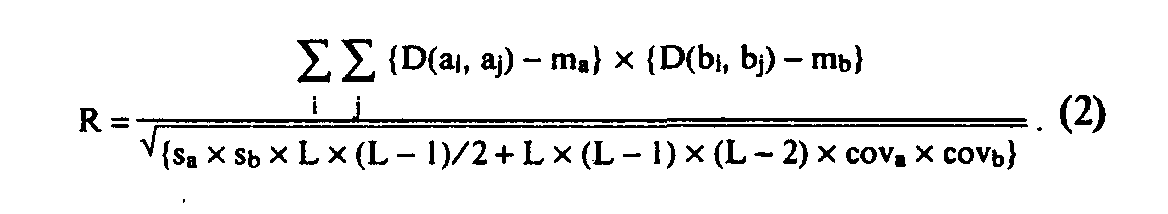
Here D is a letter proximity matrix (commonly, "letter identity matrix", used to reduce the alphabet); L is length of the motif; m, s, and cov (with indices a and b) are mean, standard deviation, and covariance of matrix D elements, taking into account frequency of letters in the first and second sequences, respectively. We called this estimate CORRELATIONAL POWER (R), analogous to estimate A from Eq. (1). Search using this criterion will probably select all strong enough motifs, no matter which matrix is used to correlate their fragments (although relatively weak motifs could be missed by this method).
The server presents motif search and alignment procedures which use both A and R correlational power criteria.
Both "homology" search procedures using A and R power consist of three stages [7]. In the first stage SHIFTS of each sequence from the database are found, matching similar fragments in query sequence (or alignment). Second, the highest similarity regions without insertions/deletions (MOTIFS) are found for each of these shifts, and

Fig. 2. Local alignment consists of two motifs (listed in uppercase). Positions with eligible substitutions, according to conventional analysis by substitution weight matrices (like the Dayhoff matrix), are marked with asterisks. Positions in each of the two motifs with the maximum contribution to correlational power of sequence fragments are marked with vertical bars (|).
then best scoring alignments are produced for a pair of sequences (or query alignment and target database sequence). If correlational power is used, then the best shifts are selected from the number of pairs of positions (i, j) in shifted sequence which follow the rule aj = aj, bj = bj (i.e., both sequences hold identical letters in this position), where distance between positions i and j is limited by some threshold. Obviously, this number well corresponds with the correlational power.
We will give an example of using the correlational power criterion. We determine motifs and produce local alignment for two protein sequences from SwissProt— trypsin TRYP_PIG and CBP2_YEAST protein—which carries out splicing of the terminal intron of cytochrome b mRNA precursor [18] (Fig. 2).
The second motif of this alignment will have very low power calculated using any of the conventional matrices (i.e., insignificant homology level). The algorithm that we propose reveals correlational link in both motifs between groups of amino acids, arranged according to equivalence of their physicochemical properties: (A, G), (D, E, N, Q), (C), (I, L, M, V), (F, Y, W), (H), (K, R), (P), (S, T). For the first motif, this link is based on identity of equivalent amino acids, while for the second one there is a correlation between specific groups of non-equivalent amino acids. The example in Fig. 2 shows that trypsin residues from (A, G) and (S, T) groups match CBP2 residues from the (I, L, M, V) group, trypsin residues from (I, L, M, V) group, as a rule, match CBP2 (S, T) residues, and residue С matches residue K. This produces natural alignments for both motifs.
Of course, if we know the desired type of residue matching in advance, we can build the best possible substitution weight scoring matrix (as described in [17]) and apply it to find the second motif from our example in a usual way. However, in the situation when the matrix is not known, but instead we are interested in all statistically significant "similarities" of sequence fragments, weighted by any matrix, this method of correlational power search has the advantage of simultaneously considering all possible matrices.
3. RNA Secondary Structure Prediction Based on Sequence Alignment
The method applied for RNA secondary structure prediction uses phylogenetic considerations for energy optimization [19]. Multiple sequence alignment, which includes the analyzed sequence, is used for reliable prediction of at least the most conserved of the secondary structure regions. The algorithm takes into account data on conservative and compensational substitutions in different positions of aligned sequences.
First, all possible complementary regions of an analyzed sequence are scanned and those exceeding the energy threshold are selected. Since the sequence is a part of an alignment, each of its complementary positions corresponds (albeit not mandatorily) to the complementary positions of all other sequences included in the alignment. If two positions are complementary in all aligned sequences, but hold different letters in different sequences, then we call this pairing complementary with COMPENSATIONAL substitutions. Complementary pairing with the same letter pairs in each of aligned positions is called CONSERVATIVE.
This method of building optimal RNA secondary structure optimizes not actual, but CONDITIONAL structure energy. This conditional energy for complete structure includes energy of both conservative and com-pensational pairing, weighted according to scoring coefficients, with the higher coefficients for the compensational substitutions. Search for optimal structure could be carried out by two methods: "greedy" full check algorithm or dynamic programming algorithm [19].
An example of the use of this procedure is presented in Fig. 3.
The method we use to predict protein secondary structure is based on the screening of a database contain ing specific profiles obtained by processing the databank of 3D structures.
During screening, the best matches between areas of the query sequence and fragments of proteins from the database of profiles are searched. Profile is defined as linear sequence of types (classes) of amino acid residue spatial arrangements inside 3D protein structure. The class of each amino acid is determined from the type of its secondary structure, residue charge, and water molecule accessibility [20].
Matches are searched by means of a preference matrix coupling residue type and its class of position in 3D protein structure [20] or using the concept of correlation for letters and classes of positions analogous to Eq. (2).
When the search is finished, the program overlays all matches found upon the analyzed sequence. The result for each type of secondary structure is a plot in which ordinate height in each position is proportional to the maximum power of the motif containing this residue.
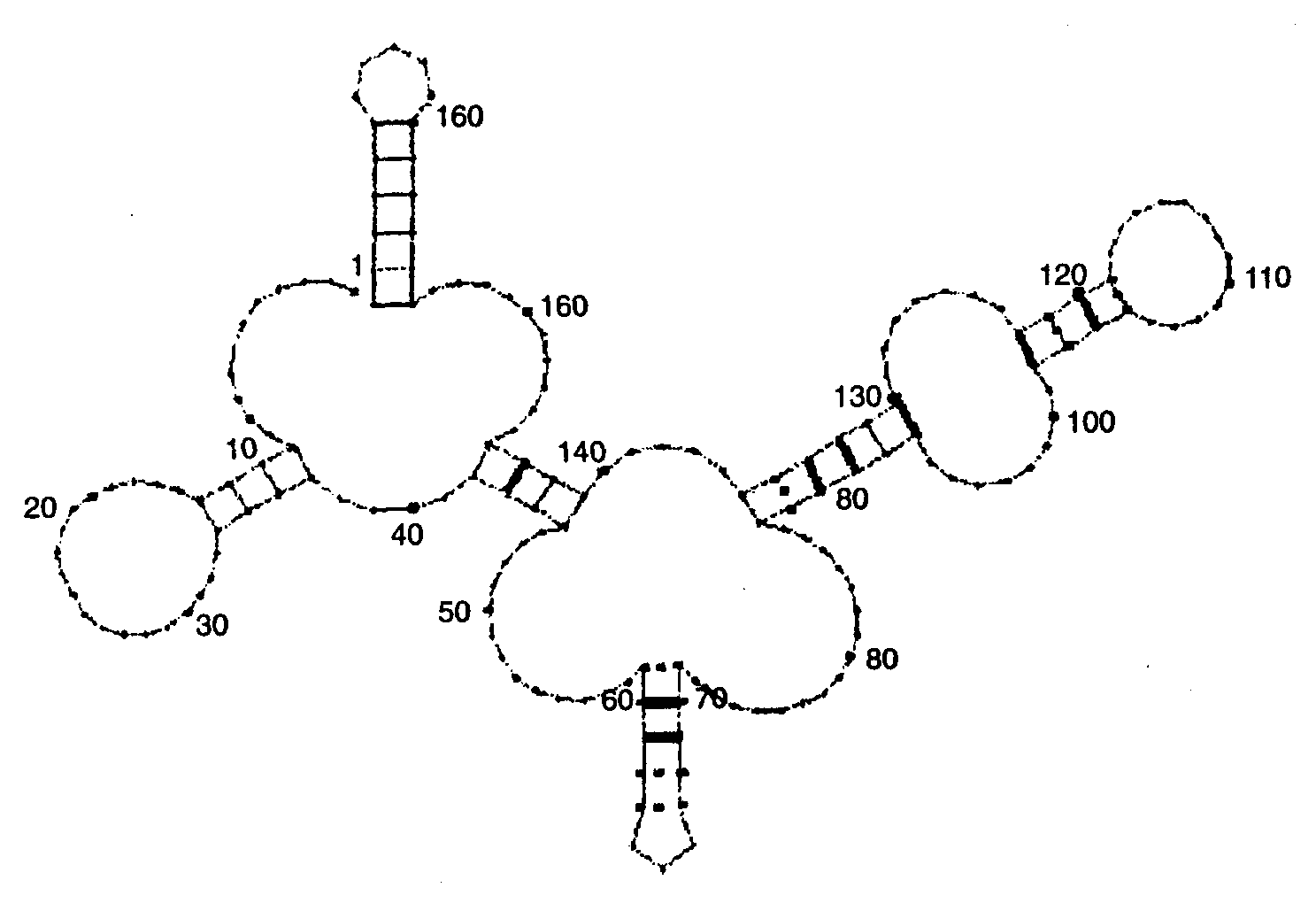
Fig. 3. Secondary structure for Q precursor of Bacillus 5S rRNA, clone p5S-A2, predicted according to the alignment of bacterial 5S rRNAs. Complementary pairs in positions with compensatory substitutions are marked with thick lines. Conservative pairs are marked with dashed lines. Nucleotide position are denoted by numbers.
5. Reverse Protein Folding
This procedure uses the idea of "similarity" between letters of protein sequence and 3D class profiles of proteins (see section 4), which is searched using both substitution weight matrix for sequence letters versus residue 3D classes [20], and correlational power, analogous to Eq. (2). Different distance matrices are used in first and second brackets while calculating correlational power according to Eq. (2): residue similarity matrix (for sequence) and residue 3D classes similarity matrix (for profile).
6. Reverse RNA Secondary Structure Folding
Locating of the nucleotide sequences in a databank with fragments similar in secondary structure to the target sequence is an important element of general "homology" search analysis. Alignment of such sequences helps to refine secondary structure prediction for this fragment by use of the compensational substitutions algorithm (see section 3) and allows to estimate conservation and functionality of the RNA secondary structure elements [21, 22].
The search algorithm is as follows: initially, RNA secondary structure is defined as a set of helices—reciprocally complementary fragments for which upper and lower neighboring distance limits are assigned. As a rule, complementary fragments have opposite orientation; however, in the case of pseudoknots the orientation may coincide. Helix element at the left side of the sequence is denoted by H with the number and length of fragment attached (e.g., Hl(7)), and the corresponding complementary fragment at the right is denoted either by R (Reverse) with the appropriate number (Rl), or by D (Direct). Hence, all secondary structure is defined as a pattern:
Hl(7) (7,8) H2(7) (4,7) D2 (9, 10) Rl, (3)
in which loop elements are separated by the limit values for the distance between their boundaries along the sequence. This notation implies equal length for elements Ri (or dj) and Hj.
Searching for this kind of putative pattern sites in the analyzed sequence is accomplished by algorithm analogous to the one used for loose matching search with the ordinary patterns (see section 1). It consists of the following steps: at the beginning, all complementary pairs (h, r) in the analyzed sequence are selected for which distances between them match the pattern of type (3). Next, couples of these pairs, (hl, rl) and (h2, r2), are examined. If this couple could be "enclosed" in the pattern, then values of the calculated function at positions (h1, r1) and (h2, r2) are incremented by one. The term "enclosed" supposes that a pair of loops (Hi, Rj) and (Hj, Rj) could be found in the pattern that satisfies the condition: if (h1, r1) and (h2, r2) are contained in (Hi, Ri) and (Hj, Rj), then the order of four letters h1, r1, h2, r2 in the sequence coincides with that of loops Hj, Ri, Hj, Rj in the pattern and distances between positions of h1, r1, h2, r2 meet the distance limits for Hi, Ri, Hj, Rj pattern.
After processing of all couples of complementary pairs of type (h1, r1) and (h2, r2) found in the sequence, we will have function F(i) (i is position number) with local minima indicating the location of putative loop sites in the target pattern (possibly, a pattern with "errors"). Then the Dothelix algorithm is applied to select sequence fragments highly matching the target secondary structure pattern.
7. Protein Structure Homology Search
Protein structure homology search (3D structure alignment) is also based on the concept of correlational power, analogous to Eq. (2):
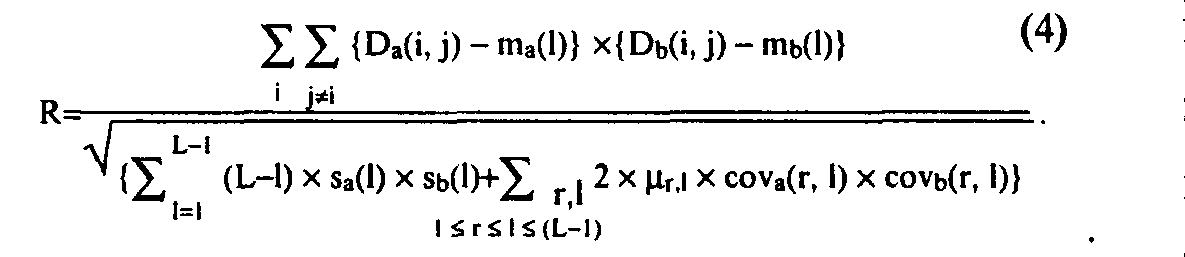
Here, D with a and b indices are matrices of spatial distance between Ca atoms inside equal in length fragments of compared 3D structures; L is length of 3D motif; m and s are mean and dispersion for elements of D matrices, depending on the distance 1 between sequence positions i and j; cov is covariance of D matrix elements, calculated separately for each set of distances 1 and r between pairs of sequence positions for which these elements are sampled; %r,l is number of motif position triplets (i, j, k) that have the distance between position i
and j equal to r, and the distance between i and k equal to 1. The main difference of Eq. (4) and Eq. (2) is that mean, dispersion, and covariance of the spatial distances between Ca atoms are assumed to depend on the interval between them in the protein sequence.
The search for fragments with similar 3D fold is performed by shifting of the protein sequences along each other. For each shift the most correlated (according to Eq. (4)) protein fragments are selected by the Dothelix algorithm.
The computer complex of the server is connected to Internet via the RADIO-MSU network (using a 115 Kbit leased line). The authors express deep gratitude to the network personnel and administration for their help in creating this server.
This work was supported in part by the Russian Foundation for Fundamental Research grant "Telecommunication Services for Genetic Information Databanks and Programs", 1994.
LITERATURE CITED
1. Gilbert, W. (1994) CABIOS, 10, 207-208.
2. Harper, R. (1994) Curr. Opinion Biotechnol., 5, 4-18.
3. Lindberg, D. (1994) Int. J. Bio-Med. Computing, 34, 13-19.
4. Appel, R. D., Bairoch, A., and Hochstrasser, D. F. (1994) Trends Biochem. Sci., 19, 258-260.
5. Devereux, J., Haeberli, P., and Smithies, O. (1984) Nucleic Acids Res., 12, 387-395.
6. Kiong, B. K., and Tan, T. W. (1993) Сотр. Appl. Biosci., 9, 211-214.
7. Brodsky, L. I., Vassilyev, A. V., Kalaydzidis, Ya. L., Osipov, Yu. S., Tatuzov, R. L., and Feranchuk, S. I. (1992) DIM ACS Series in Discrete Mathematics and Theoretical Computer Science, 8, 128-139.
8. Brodsky, L. I., Drachev, A. L., Leontovich, A. M., and Feranchuk, S. I. (1993) Biosystems, 30, 65-79.
9. Chumakov, K. M., and Yushmanov, S. V. (1988) Mol. Genet. Mikrobiol. Virusol. (Moscow), 3,3-9.
10. Yushmanov, S. V., and Chumakov, К. М. (1988) Mol. Genet. Mikrobiol. Virusol. (Moscow), 3, 9-15.
11. Bairoch, A. (1993) Nucleic Acids Res., 21, 3097-3103.
12. Leontovich, A. M., Brodsky, L. I., and Gorbalenya, A. E. (1991) Biopolimery i Kletka, 7,10-14.
13. Leontovich, A. M., Brodsky, L. I., and Gorbalenya, A. E. (1993) Biosystems, 30, 57-63.
14. Risler, J. L., Delorme, M. O., Delacroix, H., and Henaut, A. (1988) J.Mol. Вiol., 204, 1019-1029.
15. Johnson, M. S., and Overington, J. P. (1993) J. Mol. Вiol., 233, 716-738.
16. Chapman, M. S. (1994) CABIOS, 10, 111-119.
17. Leontovich, A. M. (1992) DIMACS Series in Discrete Mathematics and Theoretical Computer Science, 8, 1-9.
18. Mcgraw, P., and Tzagoloff, A. (1983) J. Biol. Chem., 258, 9459-9468.
19. Zuker, M., and Stiegler, P. (1981) Nucleic Acids Res., 9, 133-148.
20. Bowie, J. U., Luthy, R., and Eisenberg, D. (1991) Science, 253, 164-170.
21. Pilipenko, E. V., Gmyl, A. P., Maslova, S. V., Svitkin, Y. V., Sinyakov, A. N.. and Agol, V. I. (1992) Cell, 68, 119-131.
22. Shu-Yun Le, Chen, J.-H., Sonenberg, N., and Maizel, J. V. (1993) Nucleic Acids Res., 21, 2445-2451.